| Topics | |
|---|---|
| 1 | Uploading data |
| 2 | Protein inference |
| 3 | Protein inference comparison |
| 4 | Protein common names and descriptions |
The fasta file used for peptide search has to be uploaded to our protein database BEFORE you upload your search results.
You can check the fasta files available for your lab by clicking on the "Available FASTA" link in the menu.
If you do not see your file in the list please contact the administrator for uploading your fasta file.
MSDaPl supports data from two proteomic pipelines:MacCoss Lab's pipeline
Trans-Proteomic Pipeline (TPP)
There are two options for required directory structure:
Option 1:
Option 2:
The following files should be available in the experiment directory:mzXML files
pepXML files with Sequest search results. There should be one corresponding to each mzXML file.
sequest.params file used for database search
interact.pep.xml file with PeptideProphet results
interact.prot.xml file with ProteinProphet results
MSDaPl supports data from two proteomic pipelines:
Requirements for data from the MacCoss Lab's pipeline
There are two options for required directory structure:
Option 1:
Experiment directory | |---- pipeline/sequest (contains Sequest .sqt files, sequest.params and ms2 or cms2 files) | |---- pipeline/percolator (contains Percolator's .sqt files) | |---- pipeline/dtaselect/sequest (contains DTASelect-filter.txt)
Option 2:
Experiment directory | |---- sequest (contains Sequest .sqt files, sequest.params and ms2 or cms2 files) | |---- percolator (contains Percolator's .sqt files) | |---- dtaselect/sequest (contains DTASelect-filter.txt)
Requirements for data from the TPP
The following files should be available in the experiment directory:
Adding jobs to the upload queue using Web Services
MSDaPl provides REST-based web services to submit upload requests without having to use the upload form in the web interface.
In the examples below replace <server> with repoman.gs.washington.edu for MSDaPl deployed on repoman. Use flint.gs.washington.edu for the application deployed on flint.
The service provides the following REST methods:
-
Get the details of a job already in the queue
Examples using cURLURL http://<server>/msdapl_queue/services/msjob/<jobId> HTTP METHOD GET AUTHENTICATION not required PATH PRAMETER jobId PRODUCES text, xml, json
- TEXT OUTPUT: curl http://<server>/msdapl_queue/services/msjob/<jobId>
- XML OUTPUT : curl -H "Accept:application/xml" http://<server>/msdapl_queue/services/msjob/<jobId>
- JSON OUTPUT: curl -H "Accept:application/json" http://<server>/msdapl_queue/services/msjob/<jobId>
-
Get the status of a job already in the queue
Example using cURLURL http://<server>/msdapl_queue/services/msjob/status/<jobId> HTTP METHOD GET AUTHENTICATION not required PATH PRAMETER jobId
- TEXT OUTPUT: curl http://<server>/msdapl_queue/services/msjob/status/<jobId>
-
Delete a job already in the database
Example using cURLURL http://<server>/msdapl_queue/services/msjob/delete/<jobId> HTTP METHOD DELETE AUTHENTICATION required PATH PRAMETER jobId
- curl -u <username>:<password> -X DELETE http://<server>/msdapl_queue/services/msjob/delete/<jobId>
-
Submit a job to the queue
Example using cURLURL http://<server>/msdapl_queue/services/msjob/add HTTP METHOD POST AUTHENTICATION required CONSUMES text, xml, json PRODUCES text
Returns the database ID of the newly queued job
- JSON INPUT: curl -u <username>:<password> -X POST -H 'Content-Type: application/json' -d '{"projectId":"24", "dataDirectory":"/test/data", "pipeline":"MACCOSS", "date":"2010-03-29", "comments":"upload test"}' http://<server>/msdapl_queue/services/msjob/add
-
Submit a job to the queue (using query parameters)
Example using cURLURL http://<server>/msdapl_queue/services/msjob/add HTTP METHOD POST AUTHENTICATION required QUERY PRAMETERs projectId Required. ID of the parent project dataDirectory Required. path to the data directory remoteServer Optional. ID of remote server pipeline Required. Either TPP or MACCOSS date Required. Date the data was generated (Accepted format example: 09/24/10) instrument Optional. Name of the instrument use to acquire data. This should match the instruments available in MSDaPl targetSpecies Optional. Taxonomy ID of the target species comments Optional PRODUCES text
Returns the database ID of the newly queued job
- curl -u <username>:<password> -X POST "http://<server>/msdapl_queue/services/msjob/add?projectId=24&dataDirectory=/data/test&pipeline=MACCOSS&date=09/24/10&instrument=LTQ&taxId=9606&comments=some%20comments"
This document is for the protein inference program implemented in MSDaPl.
It is available for use with Percolator
results generated with the MacCoss Lab's pipeline.
The parsimonious protein inference in this program is based on the IDPicker algorithm published in:
Proteomic Parsimony through Bipartite Graph Analysis Improves Accuracy and Transparency.
Tabb et. al. J Proteome Res. 2007 Sep;6(9):3549-57
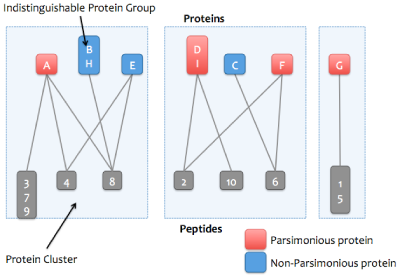
Step 1:
A bipartitie graph is created with edges between peptides and their matching proteins.
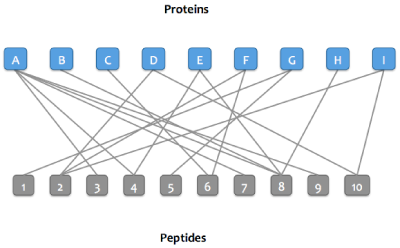
Step 2:
Peptides that match the same set of proteins are merged into a single node in the graph.
For example, peptides 3, 7, and 9 match protein A and no other protein.
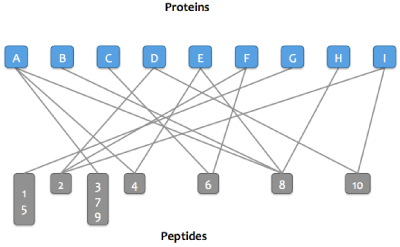
Step 3:
Proteins that match the same set of peptide are merged into a single node in the graph. These proteins comprise
an indistinguishable protein group.
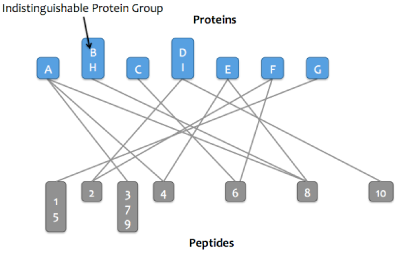
Step 4:
The graph is then resolved into its connected components, or proteins that share peptides.
Each connected component is referred to as a protein cluster.
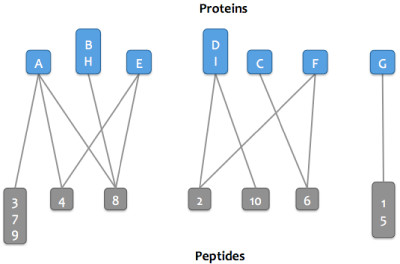
Step 5:
The smallest set of proteins sufficient to explain the peptides in each cluster are marked as parsimonious.
As of version 0.2 the protein inference program implemented in MSDaPl calculates both parsimonious and non-subset proteins. The set of parsimonious proteins is the minimum number of proteins required to explain the observed peptides. This set is always smaller (or same size) as the set of non-subset proteins (as calculated by DTASelect, for example).
In general, the number of parsimonious proteins is a good conservative estimate of the number of proteins in your sample. However, when looking for specific proteins or when compiling a list of proteins for further analysis it may be better to go with the less conservative list of non-subset proteins.
Please see the description and figures in the "Parsimonious Protein Inference" section above for information on the process of parsimonious protein inference. The figure below describes a subset protein -- a protein whose observed peptides are a subset of the observed peptides of another protein.

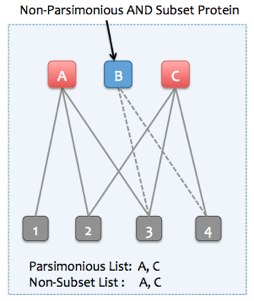
Example 1. Parsimonious protein set is the same as the non-subset protein set.
Example 2. Parsimonious protein set is the smaller than the non-subset protein set.
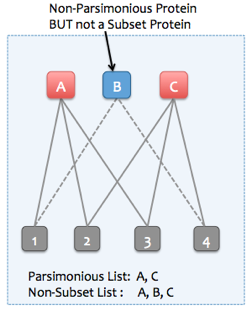
It is important to note that a parsimonious set of proteins may not be unique, as can be seen in the figure above. Proteins A and B also form a parsimonious set since they explain all the observed peptides. However, in this case, the protein that explains more observed peptides (Protein C explains 3 peptides, 1 more that protein B) is picked to be in the parsimonious set (A, C).
There can be instances where the number of peptides a protein explains is not sufficient to resolve ties, as can be seen in the example below. Any two of the three proteins (A, B, C) can be picked to form a parsimonious set. In such instances the parsimonious protein inference process makes an arbitraty choice and picks one of the three possible sets.

The protein inference view, by default displays all the inferred proteins. In order to display only the non-parsimonious proteins, check the "Exclude Non-Parsimonious" checkbox.

To display only the non-subset proteins check the "Exclude Subset" checkbox.

In order to display all proteins that were marked as non-parsimonious but were not subset proteins, check both the "Exclude Parsimonious" and "Exclude Subset" checkboxes. This will list all the proteins that would be included in a non-subset protein list but not in a parsimonious list.

Results can be filtered on q-value and Posterior Error Probability (PEP) calculated by Percolator. As of version 1.16, Percolator calculates q-values and PEP at the peptide-level in addition to scores at the PSM (Peptide Spectrum Match) level. Filters can be applied at both the peptide and PSM-level scores when inferring proteins from Percolator results where peptide-level scores are available .

Proteins (indistinguishable protein groups) can be filtered on the number of peptides and number of unique peptides identified.
The number of peptides can be calculated as one of the following:
If the "Remove Ambiguous Spectra" option is checked

any spectra that have 2 or more Percolator results that pass the q-value threshold are removed from the analysis.
Protein matches for peptides are re-calculated if the "Refresh Protein Matches" option is checked. Otherwise, protein matches reported in Sequest's SQT files are used. The protein inference process will take longer to run if this option is checked.
NOTE: Sequest may not report all protein matches for a peptide if the number of matches exceeds a hard-coded limit in Sequest. At the time of writing this documentation the limit was 21 proteins.

Isoleucine / Leucine substitutions are allowed while calculating protein matches if the second option above is checked.
Proteomic Parsimony through Bipartite Graph Analysis Improves Accuracy and Transparency.
Tabb et. al. J Proteome Res. 2007 Sep;6(9):3549-57
Parsimonious Protein Inference
Parsimonious proteins vs. Non-subset proteins
As of version 0.2 the protein inference program implemented in MSDaPl calculates both parsimonious and non-subset proteins. The set of parsimonious proteins is the minimum number of proteins required to explain the observed peptides. This set is always smaller (or same size) as the set of non-subset proteins (as calculated by DTASelect, for example).
In general, the number of parsimonious proteins is a good conservative estimate of the number of proteins in your sample. However, when looking for specific proteins or when compiling a list of proteins for further analysis it may be better to go with the less conservative list of non-subset proteins.
Please see the description and figures in the "Parsimonious Protein Inference" section above for information on the process of parsimonious protein inference. The figure below describes a subset protein -- a protein whose observed peptides are a subset of the observed peptides of another protein.
Example 1. Parsimonious protein set is the same as the non-subset protein set.
Example 2. Parsimonious protein set is the smaller than the non-subset protein set.
It is important to note that a parsimonious set of proteins may not be unique, as can be seen in the figure above. Proteins A and B also form a parsimonious set since they explain all the observed peptides. However, in this case, the protein that explains more observed peptides (Protein C explains 3 peptides, 1 more that protein B) is picked to be in the parsimonious set (A, C).
There can be instances where the number of peptides a protein explains is not sufficient to resolve ties, as can be seen in the example below. Any two of the three proteins (A, B, C) can be picked to form a parsimonious set. In such instances the parsimonious protein inference process makes an arbitraty choice and picks one of the three possible sets.
The protein inference view, by default displays all the inferred proteins. In order to display only the non-parsimonious proteins, check the "Exclude Non-Parsimonious" checkbox.
To display only the non-subset proteins check the "Exclude Subset" checkbox.
In order to display all proteins that were marked as non-parsimonious but were not subset proteins, check both the "Exclude Parsimonious" and "Exclude Subset" checkboxes. This will list all the proteins that would be included in a non-subset protein list but not in a parsimonious list.
Program Options
Protein inference implemented in MSDaPl takes Percolator
results as input.Results can be filtered on q-value and Posterior Error Probability (PEP) calculated by Percolator. As of version 1.16, Percolator calculates q-values and PEP at the peptide-level in addition to scores at the PSM (Peptide Spectrum Match) level. Filters can be applied at both the peptide and PSM-level scores when inferring proteins from Percolator results where peptide-level scores are available .
Proteins (indistinguishable protein groups) can be filtered on the number of peptides and number of unique peptides identified.
The number of peptides can be calculated as one of the following:
|
|

|
If the "Remove Ambiguous Spectra" option is checked
any spectra that have 2 or more Percolator results that pass the q-value threshold are removed from the analysis.
Protein matches for peptides are re-calculated if the "Refresh Protein Matches" option is checked. Otherwise, protein matches reported in Sequest's SQT files are used. The protein inference process will take longer to run if this option is checked.
NOTE: Sequest may not report all protein matches for a peptide if the number of matches exceeds a hard-coded limit in Sequest. At the time of writing this documentation the limit was 21 proteins.
Isoleucine / Leucine substitutions are allowed while calculating protein matches if the second option above is checked.
MSDaPl supports comparing results from two or more protein inference runs. These can be results from the protein inference
program implemented in MSDaPl and/or ProteinProphet results.
The default behavior is to include all parsimonious proteins from a dataset as well as any non-parsimonious proteins
that were inferred as parsimonious in one or more of the other datasets being compared.
(NOTE: For ProteinProphet parsimonious = NOT subsumed.) You can change this behavior by selecting one of the other two available options:

The "All" options will include all proteins from each dataset being compared.
The "Parsimonious ONLY" option will inlcude only parsimonious proteins from each dataset. This means that if a protein was parsimonious in dataset1 and non-parsimonious in dataset2, it will be listed as missing in dataset2 in the comparison analysis.
Proteins can be filtered on the accession strings in the fasta file(s) used for peptide search.
Support for filtering on common names has also been added.
Filtering criteria can either be applied to individual proteins or to protein groups
(only when "Group Indistinguishable Proteins" is checked).
 With "Keep Protein Groups" checked a protein group is filtered out of the final list only if ALL members
of the group fail to pass the filtering criteria.
With "Keep Protein Groups" checked a protein group is filtered out of the final list only if ALL members
of the group fail to pass the filtering criteria.
The results from the comparison in the figure above will be displayed as:
Options
(NOTE: For ProteinProphet parsimonious = NOT subsumed.) You can change this behavior by selecting one of the other two available options:
The "All" options will include all proteins from each dataset being compared.
The "Parsimonious ONLY" option will inlcude only parsimonious proteins from each dataset. This means that if a protein was parsimonious in dataset1 and non-parsimonious in dataset2, it will be listed as missing in dataset2 in the comparison analysis.
Comparison with indistinguishable protein groups
When comparing protein runs you can choose to group indistinguishable proteins. This is the default option.
If this option is NOT selected information about shared peptides among proteins is ignored when displaying the results.
With this option checked, proteins with identical set of peptides (indistinguishable proteins) are displayed together.
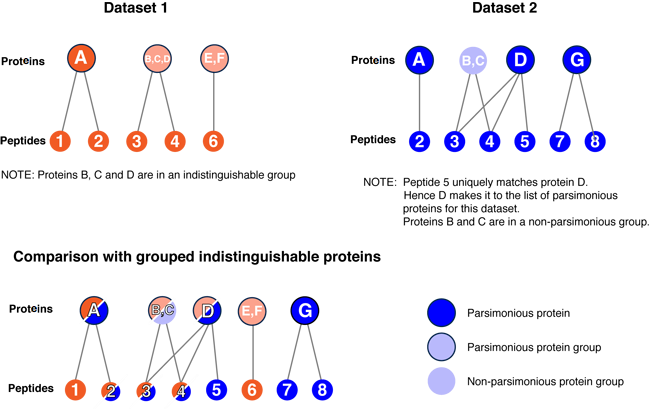
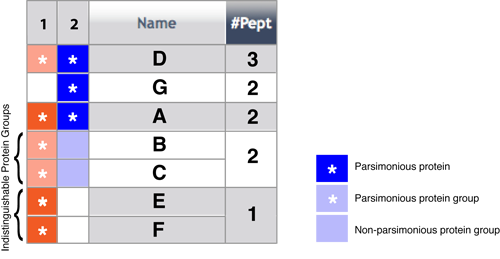
The figure below explains the process of building a list of indistinguishable protein groups from 2 datasets being compared.
The results from the comparison in the figure above will be displayed as:
Spectrum Counts
Two numbers are displayed in the spectrum counts columns for a protein in each dataset.
The first is the number of filtered (after any cutoffs applied during the protein inference process) spectra
for a protein. The second number, in parentheses, is the normalized spectrum count.
Normalization is done using the total (filtered) spectrum counts for the datasets being compared.
Frequently Asked Questions
- Q. Why is the number of proteins and protein groups displayed in the comparison view
more than the numbers in the protein inference view, or the numbers
displayed on the main project page?
A. (part 1) The number of proteins in the comparison view depends on the options used for comparison.With the default option the number of proteins included from a dataset may be more than the number of parsimonious proteins in the dataset. The default option is to select all proteins from a dataset that were either parsimonious in that dataset or one of the other datasets in the comparison analysis. Choose the "Parsinonious ONLY" option to limit the analysis to only parsimonious proteins in each dataset.
A. (part 2) The number of protein groups reported is in the context of the comparison analysis. The comparison process pools all the individual filtered proteins from each dataset and creates a bi-partite graph connecting proteins with peptides. The proteins are then grouped again into indistinguishable proteins. These groups may not be identical to those in the original datasets due to possibly different peptide identifications.
In the figure above, there were 3 protein groups in Dataset1 before comparison but 4 after comparison since one of the groups (proteins B,C,D) was split up. This happened because Dataset 2 had a unique peptide for protein D.
This document applies to the names and descriptions displayed in the protein inference and
comparision pages.
Common names are displayed only for proteins from the following supported species:
In the protein inference and comparison pages, descriptions from the fasta file used for the peptide search are also shown in addition to the best description determined above. If this description is identical to the best description it is ignored. When multiple descriptions are available for a protein, only one is shown by default. The other available descriptions can be be made visible clicking on the [+] link or the [Full Descriptions] link.
Common names are displayed only for proteins from the following supported species:
- Saccharomyces cerevisiae
- Schizosaccharomyces pombe
- Caenorhabditis elegans
- Drosophila melanogaster
- Homo sapiens
- SGD for S. cerevisiae
- Sanger Pombe for S. pombe
- WormBase for C. elegans
- HGNC (HUGO) for H. sapiens
- Swiss-Prot
- NCBI-NR
In the protein inference and comparison pages, descriptions from the fasta file used for the peptide search are also shown in addition to the best description determined above. If this description is identical to the best description it is ignored. When multiple descriptions are available for a protein, only one is shown by default. The other available descriptions can be be made visible clicking on the [+] link or the [Full Descriptions] link.